Tania Tabone,1 Richard Cotton,1 Ninan Mathew,2 J. Des Parkin,2 Judy Savige2
1Genomic Disorders Research Centre, Fitzroy, Victoria, and The University of Melbourne, Department of Medicine, Parkville, Victoria, Australia; 2The University of Melbourne, Department of Medicine (Northern Health), The Northern Hospital, Epping, Victoria, Australia
Correspondence: Judy Savige, The University of Melbourne, Department of Medicine (Northern Health), The Northern Hospital, Epping, VIC 3076, Australia. E-mail: jasavige@unimelb.edu.au
Key words: mutation detection, heteroduplex analysis, inherited disease, diagnostic assay.
Conflict of interest: the authors report no conflicts of interest.
Acknowledgements: this work was supported by the National Health and Medical Research Council of Australia and Kidney Health Australia.
Received for publication: 20 December 2009.
Revision received: 15 April 2010.
Accepted for publication: 20 April 2010.
©Copyright T. Tabone et al., 2010
Licensee PAGEPress, Italy
Journal of Nucleic Acids Investigation 2010; 1:e5
doi:10.4081/jnai.2010.e5
AbstractDirect sequencing may be problematic in demonstrating mutations where inherited disease results from multiple different heterozygous variants in large genes. We describe here a novel mutation screening method based on the ability of carbodiimide to bind mismatched DNA and interrupt primer extension thereby identifying both a heterozygous variant and its location. This assay detected all four classes of DNA mismatch in 550 bp engineered plasmid fragments and in two dominantly inherited renal diseases. In patients with thin basement membrane nephropathy, the method demonstrated multiple variants within a single amplicon including some close to the primer binding site. This method also detected a complex mutation in medullary cystic kidney disease type 2 (c.278-289 del/insCCGGCTCCT) as multiple termination events and, furthermore, correctly identified five affected and 28 unaffected family members. Carbodiimide-induced interrupted primer extension identifies heterozygous variants in large or multiexonic genes, where the variants differ in each family, their locations are unknown, and even if there are multiple known non-pathogenic variants within the same amplicon. This assay incorporates a “universal” protocol that detects all types of mutations without the need for further optimization, and potentially detects mutations where the proportion of heteroduplex is less than 50%. |
The detection of sequence variants is an important tool in diagnostic medicine. While Sanger sequencing represents the “gold standard” for mutation detection and is useful in confirming common mutations, it is expensive and may not identify previously unrecognized heterozygous changes. Thus, screening methods are often used to examine large genes where each family has a different mutation at an unknown site.
Methods for mutation detection use three main approaches: the differential hybridization of wildtype and mutant DNA as exemplified by DNA microarrays;1 changes to the physical property of mutant DNA, such as its melting profile or mobility through a gradient (high resolution melt curve analysis [hrMCA], denaturing high performance liquid chromatography [DHPLC], denaturing gradient gel electrophoresis [DGGE], and single-stranded conformational polymorphism [SSCP] analysis);2,3,4,5 and the chemical or enzymatic modification of heteroduplex DNA, such as the binding of carbodiimide or potassium permanganate, or cleavage by hydroxylamine or the plant endonuclease, CEL1.6,7,8,9,10 However, many of these methods are not suitable for routine diagnostic use because they are too insensitive or labor-intensive, involve toxic reagents, or require extensive optimization or special instrumentation. In addition, many are suitable only for DNA fragments shorter than 400 bp, and do not distinguish between a polymorphism and functional mutation occurring together within the same amplicon.
Carbodiimide is a commonly available, inexpensive, and non-toxic chemical that forms a covalent bond with the free imino moiety of thymine (T) and guanine (G) residues that are exposed in single-stranded DNA.11 This property means carbodiimide may be used to detect mutations by binding to mispaired or unpaired bases in heteroduplex DNA. Previously described carbodiimide-based mutation detection methods include retarded electrophoretic migration of bound DNA and interrupted primer extension in a gel assay.7,8 The interrupted primer extension method is based on the observation that carbodiimide binding to mismatched T and G residues inhibits DNA polymerase from extending along the DNA strand during amplification, resulting in the formation of a truncated PCR product at the site of the modified base. However, uses standardized reaction conditions for all DNA amplicons and demonstrates multiple variants simultaneously within individual amplicons.
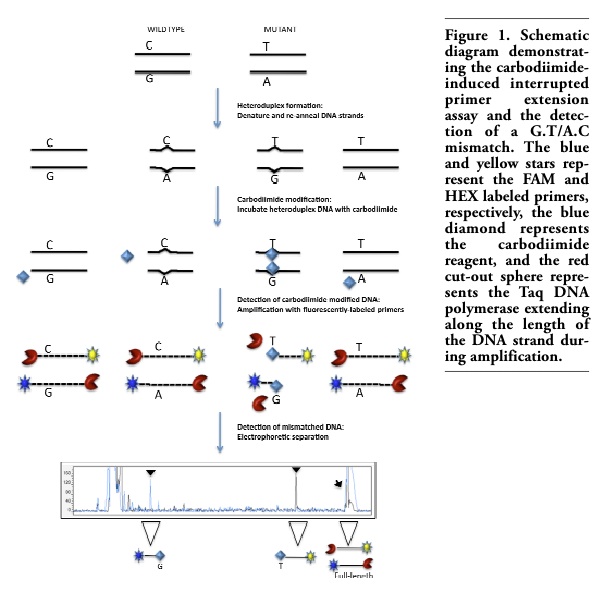
We describe here a novel modification of the carbodiimide-induced interrupted primer extension method that increases its sensitivity and throughput for mutation detection. The approach uses standardized reaction conditions for all amplicons and demonstrates multiple variants simultaneously within a single DNA amplicon. It not only confirms the presence of a mutation within an individual amplicon but also indicates its exact location in the same reaction (Figure 1).
The method is used here to detect point and insertion/deletion (INDEL) mutations and polymorphisms in two common dominantly inherited kidney diseases: thin basement membrane nephropathy (MIM 141200) and medullary cystic kidney disease type 2 (MIM 603860). Thin membrane nephropathy affects at least 1% of the population, and is the most common cause of persistent glomerular hematuria worldwide.12,13,14 It is a result of mutations in the COL4A3 and COL4A4 genes, which encode the α3 and α4 chains of type IV collagen, the major constituent of the glomerular basement membrane.15 Functional mutations in thin basement membrane nephropathy are different in each family, and are usually single nucleotide changes distributed randomly along these genes. Medullary cystic kidney disease type 2 is characterized by progressive renal impairment, kidney cysts, and sometimes hyperuricemia. It results from mutations in the gene encoding Tamm-Horsfall/uromodulin (UMOD), the most abundant protein found in the urine.16 The majority of mutations identified in the UMOD gene to date cluster in exons 4 and 5.17
pBR322 plasmid clones engineered with one of eight different single point mutations in the β-globin promoter region18 were used to assess the ability of the method to detect each class of mismatch, as well as mismatches at various distances from the primer binding site (Table 1).
The interrupted primer extension method was studied for its ability to detect a range of mutations in different sequence contexts in human disease and to compare its performance with established mutation detection methods. Genomic DNA samples were examined from 37 patients with biopsy-proven thin basement membrane nephropathy that were all screened previously by SSCP for mutations in exon 25 of the COL4A3 gene. In addition, DNA samples were de-identified and screened from 33 adult members of a family with medullary cystic kidney disease type 2 of whom five had typical clinical features (renal failure, renal cysts on imaging or renal biopsy) and from 40 healthy blood donors for mutations in exon 4 of UMOD. All the results were validated by Sanger sequencing (Australian Genome Research Facility). This study had the approval of the Human Research Ethics Committee of Austin Health and all participants provided signed informed consent.
Plasmid and genomic DNA was amplified in a 20 μL reaction volume comprising 100 ng template DNA, 0.2 mM of each dNTP (Bioline), 1.5 mM MgCl2, 1X PCR buffer (Invitrogen), 0.3 μM of each primer (Sigma-Genosys) (Table 1), and 1 U of Taq DNA polymerase (Invitrogen). The cycling conditions for all amplicons were as follows: an initial denaturation at 95°C for 5 min, followed by 35 cycles of heating to 95°C for 20 sec, annealing at the temperature optimal for the primer set (Table 1) for 20 sec, and extension at 72°C for 20 sec.
Following PCR amplification, DNA samples were denatured at 95°C for 10 min, and cooled from 80-25°C, decreasing by 1°C every 2 min in a thermal cycler to promote heteroduplex formation. To detect homozygous sequence variants, PCR products were mixed with equal volumes of wildtype PCR product in a 20 μL reaction volume before initiating heteroduplex formation. The wildtype product was generated from DNA from a non-hematuric, non-proteinuric person who had no pathogenic variants identified on DNA sequencing and in whom all non-pathogenic variants were known.
Ten microliters (0.5-1.5 μg) of crude PCR product were incubated with 50 mM N-cyclohexyl-N’-(2-morpholinoethyl) carbodiimide methyl-p-toluenesulfonate (Fluka) and 0.1 M sodium borate, pH 8.0 (Sigma) in a 40 μL ’reaction volume at 30°C for 1 h. The reaction was quenched and excess carbodiimide removed using a QIAquick PCR purification column (Qiagen) according to the manufacturer’s instructions except that isopropanol was substituted for ethanol in Buffer PB. The carbodiimide-bound DNA was eluted in 30 μL dH2O and used as a template in a second amplification round.
This reaction comprised 5 μL carbodiimide-treated DNA, 0.1 mM of each dNTP, 1.5 mM MgCl2, 1x PCR buffer, 0.1M 6-FAM labeled forward primer, 0.1 μM HEX-labeled reverse primer, and 0.25 U of Taq DNA polymerase in a 10 μL reaction volume.19 The amplification consisted of a single cycle with denaturation at 95°C for 5 min, annealing at 50°C for 1 min, and extension at 72°C for 5 min. The products were then denatured at 95°C for 2 min, plunged onto ice and subjected to electrophoretic separation in a 4.25% polyacrylamide gel (39:1) at 70W for 3 hr on an ABI377 Genetic Analyzer (Applied Biosystems). This method was expected to detect all single-base mutations since each mismatch class generated at least one unpaired G or T in heteroduplex DNA (G.A/T.C, A.A/T.T, C.C/G.G, and G.T/A.C).
pBR322 plasmid clones engineered with eight different mutations in the β-globin promoter sequence were used to validate the sensitivity and specificity of the interrupted primer extension assay to detect all four classes of mismatch (G.A/T.C, A.A/T.T, C.C/G.G, and G.T/A.C). Truncated amplification fragments were detected for each mismatch class and were readily distinguished from the wildtype PCR products (Figure 2A-E). ’For each mismistch class, the number of peaks of expected size (Table 1) were found (Table 1) were found and no “false positives” were detected. An example and interpretation for each mismatch class is provided. The G.A/T.C mismatch resulted in carbodiimide-bound G and T residues in the same strand orientation, forming a single termination peak as a result of truncated amplification by the forward primer (Figure 2B). The A.A/T.T mismatch resulted in carbodiimide binding to the mismatched T residues on alternative DNA strands, which were amplified by both the forward and reverse primers and hence resulted in two different sized PCR products (Figure 2C). Similarly, the C.C/G.G mismatch resulted in mismatched G residues on alternative strands, forming two differently sized truncated products as a result of carbodiimide binding to both G residues (Figure 2D). The G.T/A.C mismatch resulted in mismatched G and T residues on alternative strands that underwent carbodiimide modification producing two termination peaks of different lengths as a result of the truncated amplification from both primers (Figure 2E). The use of dual-labeled forward and reverse primers confirmed the mutations in both directions and enabled the detection of an A>C mutation by detecting the complementary G>T mutation on the opposite strand.
The carbodiimide-induced interrupted primer extension method confirmed the presence of two heterozygous mutations (c.1594G>T and c.1750G>T) and a homozygous polymorphism (c.1721C>T) in exon 25 of the COL4A3 gene in three individuals from the 37 with thin basement membrane nephropathy who were studied. The interrupted primer extension method detected all mutations identified with SSCP and did not detect any novel changes.
The c.1594G>T and c.1750G>T mutations both resulted in G.A/T.C mismatches (Table 1). The carbodiimide-bound G and T residues were in the anti-sense orientation and were
only detected by the forward primer. The resulting truncated product for c.1750G>T was 31 bp in length but still readily distinguishable from unincorporated primers, dNTPs, and primer-dimer (Figures 3A and B). The c.1721T>C polymorphism resulted in a G.T/A.C mismatch that produced termination products that were detected in both directions (Table 1).
To assess the interrupted primer extension method in a clinical setting, 33 DNA samples from a family with medullary cystic kidney disease type 2 and 40 control DNA samples were de-identified and screened for mutations. Analysis of the 73 DNA samples in a blinded manner allowed an unbiased determination of the sensitivity and specificity of the assay for mutation detection ’as well as evaluation of the ease of interpreting chromatograms without having any prior genotype knowledge. The interrupted primer extension method identified a complex heterozygous mutation in exon 4 of the UMOD gene in five family members diagnosed with medullary cystic kidney disease type 2 clinically and with imaging (Figures 3C and D). The variant was not present in the 28 unaffected family members or any of the 40 control DNA samples. Sanger sequencing was performed to characterize the mutation and further validated these findings. The UMOD gene mutation (c.278-289 del/insCCGGCTCCT) was a 12 bp deletion with a 9 bp insertion. After heteroduplex formation, this mutation resulted in multiple mispaired and ’unpaired G and T bases producing many termination fragments with both the forward and reverse primers, which were detected as a series of termination peaks on electrophoretic separation. Figure 3E shows a theoretical schematic of the potential carbodiimide binding sites generated after heteroduplex formation of the insertion/deletion mutation that would result in a series of mispaired and unpaired bases.
Carbodiimide-induced interrupted primer extension is a sensitive and robust assay for mutation scanning in inherited disease. It reduces the amount of sequencing required by accurately locating the DNA mismatch, identifies pathogenic mutations even when another variant is present simultaneously within the amplicon, and differentiates between pathogenic and known non-pathogenic variants.
The studies described here demonstrated that interrupted primer extension detected all four classes of mismatch in a 550 bp PCR product from plasmid DNA samples. Furthermore each assay used the same conditions; in particular, reagent concentrations and incubation temperatures. This assay detected all 12 mutations from a total of 119 DNA samples, and the results with interrupted primer extension were concordant with those from SSCP in thin basement membrane nephropathy and with the clinical and histological diagnosis of medullary cystic kidney disease in members of a large affected family.
Carbodiimide is a widely-available, inexpensive, and safe laboratory reagent. The interrupted primer extension assay was simple to perform, and chromatograms were interpreted easily. The use of universal M13 fluorescent primers17 in the second amplification round provided the opportunity for multiplexing and electrophoretic separation, and helped reduce costs. The standardized conditions were compatible with robotic automation and high-throughput set-ups.
The novel modifications described here addressed the issues that limited sensitivity in previously described carbodiimide-based mutation detection methods.7,8 The sensitivity of detecting mismatched DNA was enhanced with the use of fluorescent- rather than radio-labeled DNA. The DNA yield itself was increased with the use of fluorescent-labeled DNA, elimination of a precipitation step by incubating the crude PCR product with carbodiimide excess, and precipitation of the DNA with isopropanol because carbodiimide is soluble in ethanol. The modifications also enhanced the method’s specificity. Firstly, the use of a high salt concentration buffer (100 mM sodium borate) facilitated compression of the duplex DNA and potentially reduced “breathing” of the DNA ends thereby minimizing non-specific binding with internal G and T bases. This allowed the use of excess carbodiimide in the assay to ensure sufficient binding to all classes of mismatched DNA, especially G.G/C.C where binding is weakest.7 Secondly, reducing the concentration of PCR primers and DNA polymerase in the second amplification round meant the chromatograms were easier to interpret and also minimized the yield of non-specific product.
Carbodiimide-induced interrupted primer extension represents a novel approach for mutation detection in large, multi-exonic genes where the pathogenic mutations are different in each family, their locations are unknown, and they must be distinguished from multiple known non-pathogenic variants within the same amplicon. The assay described here incorporates a universal protocol that detects all types of mutations and eliminates reaction optimization for individual amplicons. This method may be particularly useful in detecting mutations where the proportion of heteroduplex is less than 50%.
|
Figure 1. Schematic diagram demonstrating the carbodiimide-induced interrupted primer extension assay and the detection of a G.T/A.C mismatch. The blue and yellow stars represent the FAM and HEX labeled primers, respectively, the blue diamond represents the carbodiimide reagent, and the red cut-out sphere represents the Taq DNA polymerase extending along the length of the DNA strand during amplification. |
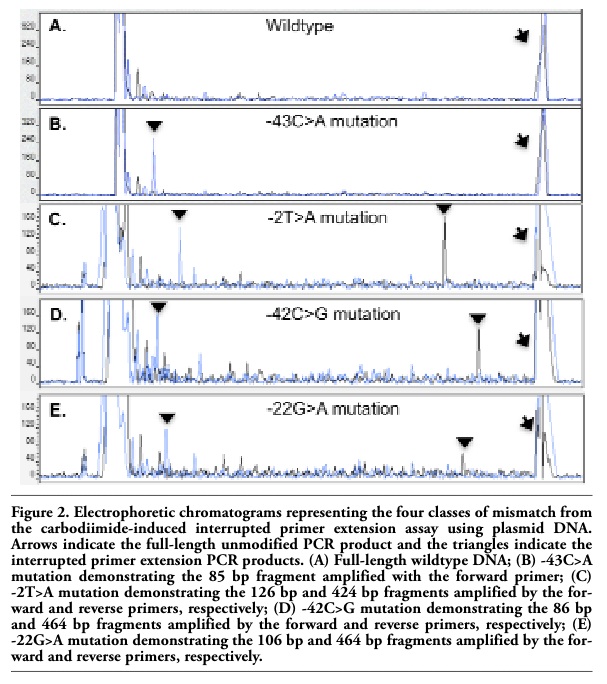 |
Figure 2. Electrophoretic chromatograms representing the four classes of mismatch from the carbodiimide-induced interrupted primer extension assay using plasmid DNA. Arrows indicate the full-length unmodified PCR product and the triangles indicate the interrupted primer extension PCR products. (A) Full-length wildtype DNA; (B) -43C>A mutation demonstrating the 85 bp fragment amplified with the forward primer; (C) -2T>A mutation demonstrating the 126 bp and 424 bp fragments amplified by the forward and reverse primers, respectively; (D) -42C>G mutation demonstrating the 86 bp and 464 bp fragments amplified by the forward and reverse primers, respectively; (E) -22G>A mutation demonstrating the 106 bp and 464 bp fragments amplified by the forward and reverse primers, respectively. |
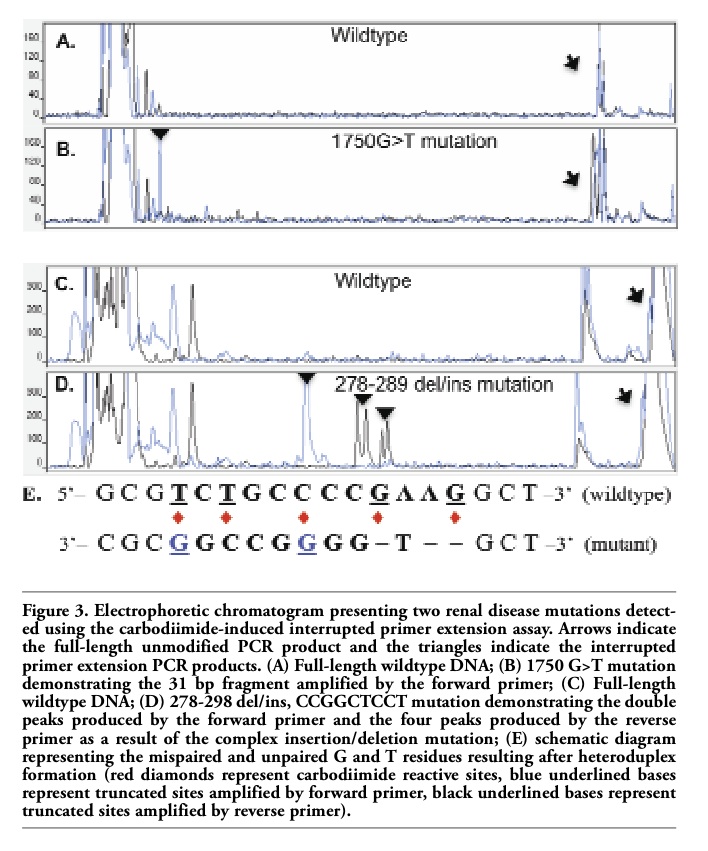 |
Figure 3. Electrophoretic chromatogram presenting two renal disease mutations detected using the carbodiimide-induced interrupted primer extension assay. Arrows indicate the full-length unmodified PCR product and the triangles indicate the interrupted primer extension PCR products. (A) Full-length wildtype DNA; (B) 1750 G>T mutation demonstrating the 31 bp fragment amplified by the forward primer; (C) Full-length wildtype DNA; (D) 278-298 del/ins, CCGGCTCCT mutation demonstrating the double peaks produced by the forward primer and the four peaks produced by the reverse primer as a result of the complex insertion/deletion mutation; (E) schematic diagram representing the mispaired and unpaired G and T residues resulting after heteroduplex formation (red diamonds represent carbodiimide reactive sites, blue underlined bases represent truncated sites amplified by forward primer, black underlined bases represent truncated sites amplified by reverse primer). |
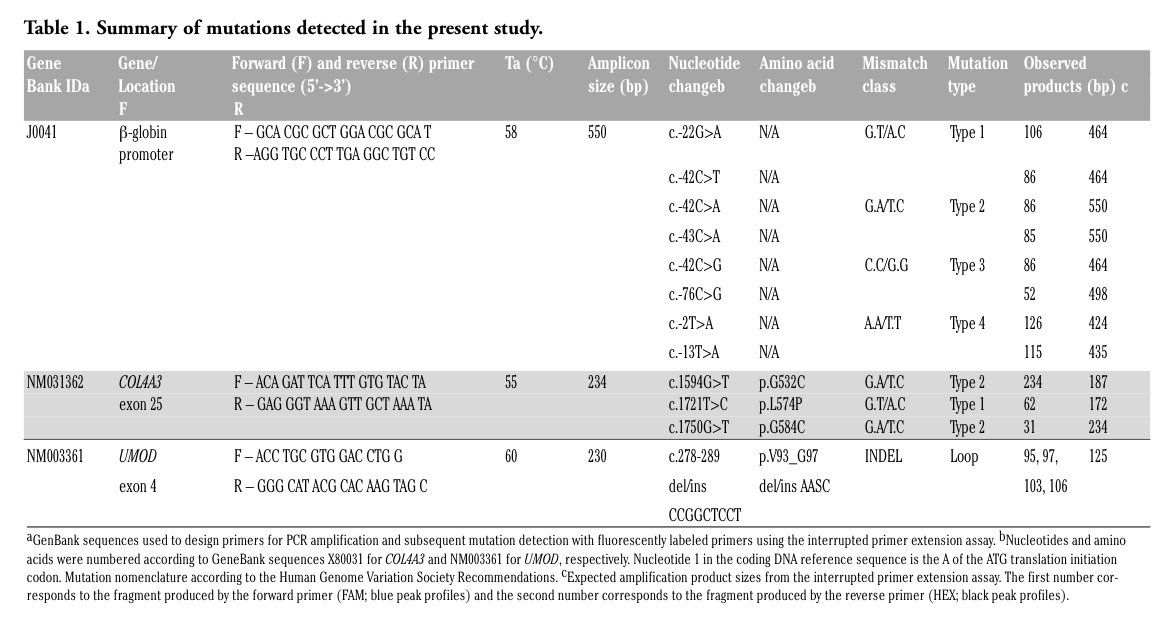 |
Table 1. Summary of mutations detected in the present study. |
1. Beaudet AL, Belmont JW. Array-based DNA diagnostics: Let the revolution begin. Annu Rev Med 2008;59:113-29. [PubMed]
2. Myers RM, Maniatis T, Lerman LS. Detection and localization of single base changes by denaturing gradient gel electrophoresis. Methods Enzymol 1987;155: 501-27. [PubMed]
3. Oefner PJ, Underhill PA. Comparative DNA sequencing by denaturing high-performance liquid chromatography (DHPLC). Am J Hum Genet 1995;57: A266.
4. Orita M, Iwahana H, Kanazawa H, et al. Detection of polymorphisms of human DNA by gel electrophoresis as single-strand conformation polymorphisms. Proc Natl Acad Sci USA 1989;86:2766-70. [PubMed]
5. Wittwer CT, Reed GH, Gundry CN, et al. High-resolution genotyping by amplicon melting analysis using LCGreen. Clin Chem 2003;49:853-60. [PubMed]
6. Cotton RG, Rodrigues NR, Campbell RD. Reactivity of cytosine and thymine in single-base-pair mismatches with hydroxylamine and osmium tetroxide and its application to the study of mutations. Proc Natl Acad Sci USA 1988;85:4397-401. [PubMed]
7. Ganguly A, Prockop DJ. Detection of single-base mutations by reaction of DNA heteroduplexes with a water-soluble carbodiimide followed by primer extension: application to products from the polymerase chain reaction. Nucleic Acids Res 1990;18: 3933-9. [PubMed]
8. Novack DF, Casna NJ, Fischer SG, et al. Detection of single base-pair mismatches in DNA by chemical modification followed by electrophoresis in 15% polyacrylamide gel. Proc Natl Acad Sci USA 1986;83:586-90. [PubMed]
9. Oleykowski CA, Bronson Mullins CR, Godwin AK, et al. Mutation detection using a novel plant endonuclease. Nucleic Acids Res 1988;26:4597-602. [Abstract]
10. Tabone T, Sallmann G, Webb E, et al. Detection of 100% of mutations in 124 individuals using a standard UV/Vis microplate reader: a novel concept for mutation scanning. Nucleic Acid Res 2006; 34:e45. [PubMed]
11. Lebowitz J, Garon CG, Chen MC, et al. Chemical modification of simian virus 40 DNA by reaction with a water-soluble carbodiimide. J Virol 1976;18:205-10. [PubMed]
12. Buzza M, Wang YY, Dagher H, et al. COL4A4 mutation in thin basement membrane disease previously described in Alport syndrome. Kidney Int 2001;60:480-3. [PubMed]
13. Lemmink HH, Nillesen WN, Mochizuki T, et al. Benign familial hematuria due to mutation of the type IV collagen alpha-4 gene. J Clin Invest 1996;98:1114-8. [PubMed]
14. Savige J, Rana K, Tonna S, et al. Thin basement membrane nephropathy. Kidney Int 2006;64:1169-78.
15. Wang YY, Rana K, Tonna S, et al. COL4A3 mutations and their clinical consequences in thin basement membrane nephropathy (TBMN). Kidney Int 2004;65:786-90. [PubMed]
16. Hart TC, Gorry MC, Hart PS, et al. Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricemia nephropathy. J Med Genet 2002;39:882-92. [Abstract]
17. Wolf MT, M ucha BE, Attanasio M, et al. Mutations of the uromodulin gene in MCKD2 patients cluster in exon 4, which encodes 3 EGF-like domains. Kidney Int 2003;64:1580-7. [PubMed]
18. Myers RM, Larin Z, Maniatis T. Detection of single base substitution by ribonuclease cleavage at mismatches in RNA:DNA duplexes. Science 1985;230:1242-6. [Abstract]
19. Schuelke M. An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol 2000;18:233-4. [PubMed]
[TOP]